- Wprowadzenie
- Materiały i metody
- Uczestnicy
- Standaryzacja diety
- Protokół infuzji
- Zbieranie próbek krwi
- Zbieranie próbek moczu
- Metoda analityczna
- Bezpieczeństwo
- Analiza statystyczna
- Etyka
- Wyniki
- Bezpieczeństwo
- Osocze
- Mocz
- Dyskusja
- Dostępność danych
- Oświadczenie o etyce
- Wkład autorów
- Funding
- Oświadczenie o konflikcie interesów
- Podziękowania
Wprowadzenie
Nukleotyd macierzysty pirydyny, dinukleotyd nikotynamidowo-adeninowy (NAD+) jest obecny we wszystkich komórkach ciała i jest niezbędny dla żywotności i funkcji komórek.
Jako kofaktor odpowiedzialny za transport elektronów do łańcucha oddechowego, NAD+ i jego para redoks NADH są kluczowe dla produkcji energii (ATP) w mitochondriach poprzez fosforylację oksydacyjną. Fosforylowany metabolit NAD+, NADP+ z jego parą redoks NADPH, dostarcza również siły redukującej do wielu reakcji anabolicznych, w tym syntezy cholesterolu i kwasów nukleinowych, wydłużania kwasów tłuszczowych i regeneracji glutationu (GSH), jednego z głównych antyoksydantów organizmu. W sumie ta rodzina nukleotydów pirydynowych przyczynia się do wymiany redoks w ponad 400 reakcjach enzymatycznych. Co ważne, podczas działania jako para redoks NAD+ nie jest zużywany. Jednakże, NAD+ służy również jako substrat dla wielu innych ważnych procesów metabolicznych i dlatego jest zużywany w wyniku ich reakcji chemicznych, potencjalnie wyczerpując zasoby NAD+ w tkankach. Do tej liczby należą reakcje napędzane przez rodzinę enzymów poli adenozyno-fosforybozy-rybozy (ADPR) (PARP 1-17) kontrolujących naprawę DNA i stabilność jądrową, enzymy kontroli epigenetycznej (Sirt1-7), międzykomórkową komunikację immunologiczną (CD38/CD157) i regenerację neuronów (SARM1; Essuman i in., 2017). Potencjał istnieje zatem dla zaburzeń metabolizmu komórkowego na wielu poziomach w warunkach, w których zużycie NAD+ przez te enzymy przekracza podaż NAD+ lub syntezę.
Kliniczne znaczenie utrzymania komórkowych poziomów NAD+ zostało ustalone na początku ubiegłego wieku wraz z odkryciem, że pellagra, choroba charakteryzująca się biegunką, zapaleniem skóry, demencją i śmiercią, może być wyleczona za pomocą żywności zawierającej prekursor NAD+ – niacynę (znaną również jako witamina B3; Goldberger, 1914). Chociaż pellagra jest rzadka w krajach rozwiniętych, wykazano, że komórkowe stężenie NAD+ zmniejsza się w warunkach zwiększonego uszkodzenia oksydacyjnego, jakie występuje podczas starzenia się (Braidy et al., 2011; Massudi et al., 2012; Guest et al., 2014). Altered levels of NAD+ have been found to accompany several disorders associated with increased oxidative/free radical damage including diabetes (Wu et al., 2016), heart disease (Pillai et al., 2005), age-related vascular dysfunction (Csiszar et al., 2019), ischemic brain injury (Ying and Xiong, 2010), misfolded neuronal proteins (Zhou et al., 2015) i demencji Alzheimera (Abeti i Duchen, 2012).
Oprócz patologicznych znaków rozpoznawczych, jakimi są blaszki Aβ i splątki neurofibrylarne tau, uszkodzenia oksydacyjne są spójnym znaleziskiem w chorobie Alzheimera i są powszechnie uznawane za wczesne wydarzenie w procesie patogenetycznym, nawet poprzedzające odkładanie się Aβ (Su i in., 2008). Chociaż reaktywne formy tlenu (ROS) powodujące to uszkodzenie prawdopodobnie pochodzą z wielu źródeł, uważa się, że dominującą rolę odgrywają dysfunkcyjne mitochondria i dostępność metali aktywnych redoks, takich jak Fe++ i Cu+ (Zhua i in., 2007).
Główną konsekwencją komórkowego stresu oksydacyjnego, w mózgu i w innych miejscach, są jedno- lub dwuniciowe pęknięcia DNA. W odpowiedzi na uszkodzenia DNA, PARP1 hydrolizuje NAD+ w celu wytworzenia polimerów ADP-rybozy (Ying, 2013). Wykazaliśmy wcześniej, że poziomy NAD+ były odwrotnie skorelowane z miarami stresu oksydacyjnego w tkankach ludzkich (Massudi i in., 2012) oraz w mózgu szczura (Braidy i in., 2014). Tak więc, podczas gdy obniżony poziom NAD+ w mózgu żyjących osób cierpiących na chorobę Alzheimera czeka na potwierdzenie technikami nieinwazyjnymi, spójne odkrycia uszkodzeń oksydacyjnych w mózgu post mortem silnie wspierają pogląd, że przyspieszony obrót NAD+ i wyczerpanie przyczyniają się do dysfunkcji neurologicznej w tej chorobie. Ponieważ interwencje ukierunkowane na przywrócenie NAD+ wykazano w modelach zwierzęcych w celu wspierania zdrowego starzenia się i poprawy funkcji metabolicznych (Yoshino i in., 2011; Mills i in., 2016) i demencji (Long i in., 2015), strategie mające na celu podniesienie poziomu NAD+ u ludzi są aktywnie explored.
Najbardziej bezpośrednią metodą zwiększenia poziomu NAD+ jest dożylne (IV) podawanie. Chociaż dane z badań eksperymentalnych są minimalne, znaczące korzyści kliniczne z IV infuzji NAD+ w wycofaniu alkoholu zostały wcześniej zgłoszone (O’Holleran, 1961; Mestayer, 2019). Co zaskakujące, podczas gdy doustne podawanie prekursorów NAD+, takich jak rybozyd nikotynamidu (NR) lub mononukleotyd nikotynamidu (NMN), jest entuzjastycznie badane pod kątem ich wpływu na poziom NAD+ (Yoshino i in., 2011, 2018; Mills i in., 2016; Airhart i in., 2017), losy metaboliczne i właściwości farmakokinetyczne dożylnego podawania NAD+ nie zostały jeszcze zgłoszone u ludzi. W związku z tym w niniejszym badaniu po raz pierwszy przedstawiono zmiany w stężeniach NAD+ i jego metabolitów podczas dożylnego wlewu NAD+ w kohorcie zdrowych uczestników płci męskiej.
Materiały i metody
Uczestnicy
Jedenastu (Test n = 8, Kontrola n = 3) uczestników płci męskiej w wieku 30-55 lat rekrutowano poprzez ogłoszenia w radiu, telewizji i mediach społecznościowych. Wszyscy uczestnicy mieli BMI poniżej 30 kg/m2 (Test średnia BMI = 27,5 ± 2,5 kg/m2; Kontrola średnia BMI = 24,6 ± 6,5 kg/m2), nie chorowali na cukrzycę, palili mniej niż 1 papierosa i spożywali mniej niż 2 standardowe napoje alkoholowe dziennie. Z badania wykluczono osoby przyjmujące leki obniżające stężenie lipidów lub przeciwzapalne, z niewydolnością wątroby lub nerek w wywiadzie, z niedawno przebytą infekcją bakteryjną, urazem lub innymi znaczącymi lub nieleczonymi zaburzeniami medycznymi. Uczestnicy otrzymali wynagrodzenie pieniężne za swój czas i udział w badaniu, niezależnie od tego, czy ukończyli eksperyment. Uczestnikom nie wolno było stosować naturalnych produktów zdrowotnych zawierających NAD+, NR lub nikotynamid w ciągu 14 dni przed badaniem i w jego trakcie.
Standaryzacja diety
W dniu poprzedzającym wlew dożylny NAD+ uczestnicy spożywali identyczną dietę o obniżonej zawartości niacyny i pili tylko wodę.
W dniu badania nie spożywano żadnych pokarmów, aż do momentu pobrania końcowej próbki krwi/moczu po 8 godzinach. Uczestnicy byli zachęcani do picia wody, aby pozostać normalnie nawodnionymi. Żadne inne rodzaje napojów nie były dozwolone.
Aby zapewnić uczestnikom odpowiednie spożycie energii podczas 6-godzinnej infuzji, roztwór dożylny zawierał również 0,1% dekstrozy, dostarczając około 2000 kalorii w ciągu 6-godzinnego okresu infuzji.
Protokół infuzji
Uczestników randomizowano do grupy badanej (n = 8) lub kontrolnej (n = 3). Uczestnikom w grupie badanej podawano dożylnie 750 mg NAD+ w normalnej soli fizjologicznej (Archway Apothecary, Covington, LA, USA) przez okres 6 godzin (szybkość infuzji = ~2 mg/min ≡ 3 μmoles/min). Ta dawka NAD+ została ustalona empirycznie i odzwierciedla schemat dawkowania powszechnie stosowany w klinikach (np. Springfield Wellness Clinic, Springfield, LA, USA), które w praktyce klinicznej regularnie podają dożylne wlewy NAD+. Uczestnikom w grupie kontrolnej podawano dożylnie zwykłą sól fizjologiczną przez okres 6 h.
Kliniczna administracja i nadzór nad wlewami dożylnymi i zbieraniem próbek zostały przeprowadzone w Springfield Wellness Center, Springfield, LA, USA.
Zbieranie próbek krwi
Próbki krwi linii podstawowej (TO) zostały pobrane u wszystkich uczestników po nocnym 12-godzinnym poście tak, aby wystąpiły bezpośrednio przed rozpoczęciem wlewu. Dodatkowe próbki pobierano następnie w 30, 60, 120 (2 h), 360 (6 h) i 480 (8 h) minutach po rozpoczęciu wlewu.
Krew pełną pobierano poprzez standardowe nakłucie żyły (ramię przeciwne do miejsca wlewu) do 5 mL nieżelowej heparynizowanej probówki. Natychmiast po pobraniu, krew odwirowano w temperaturze 4°C przez 10 minut przy 1,409× g.
Frakcje osocza i czerwonych krwinek zostały natychmiast oddzielone i rozdzielone do 5 × 500 μL alikwotów każda. Wszystkie podwielokrotności zostały następnie natychmiast zamrożone i przechowywane w temperaturze -80°C do czasu analizy.
Zbieranie próbek moczu
Po zebraniu podstawowej próbki moczu ze środkowego strumienia, uczestnicy zostali poproszeni o oddanie całego moczu do pojemnika(ów) dostarczonego(ych) w ciągu 30 min, 2 h, 6 h i 8 h po rozpoczęciu wlewu NAD+. Jeśli uczestnicy musieli oddawać mocz z przerwami pomiędzy tymi punktami czasowymi, byli proszeni o oddanie go do kolejnego pojemnika. Wszystkie próbki były alikwotowane i przechowywane w temperaturze -80°C natychmiast po otrzymaniu.
Metoda analityczna
Chromatograficzna separacja NAD+ i powiązanych metabolitów oraz detekcja MS Chromatografia cieczowa sprzężona z tandemową spektrometrią mas (LC/MS/MS) została przeprowadzona przy użyciu spektrometru mas Sciex QTRAP 5500 (Sciex, Redwood City, CA, USA), jak wcześniej opisano (Clement i in., 2018). Krótko mówiąc, 100 μL ludzkiego osocza lub moczu ekstrahowano w 400 μL lodowatego metanolu, odwirowywano w temperaturze 4°C przez 10 min i filtrowano przez wkłady membranowe 3 kDa. Ekstrakty próbek suszono w próżni, rekonstytuowano w 200 μL buforu 100 mM NH4OAc i przenoszono do szklanych fiolek o pojemności 200 μL i zakrywano przed analizą LC/MS/ MS. Standardy i próbki (20 μL) wstrzykiwano na kolumnę Phenomenex NH2 (150 mm- 2 mm- 3 mm), jak opisano wcześniej. Użyto gradientu dwuskładnikowego rozpuszczalnika składającego się z 5 mM NH4OAc pH 9,5 skorygowanego amoniakiem (faza ruchoma A) i acetonitrylu (faza ruchoma B) z szybkością przepływu 250 μL/min. Początkowy skład rozpuszczalnika podczas wstrzykiwania wynosił 25 % A, po czym następował 2-minutowy gradient do 45 % A oraz szybki wzrost gradientu do 80 % A (0,1 min), który utrzymywano przez 5,9 min, ponownie zwiększano A do 95 % (2 min), utrzymywano przez 13 min, a następnie powracano do warunków początkowych (0,1 min) w celu wyrównania, przy czym całkowity czas trwania badania wynosił 30 min. Przepływ w kolumnie kierowany był do detektora MS. Krzywe kalibracji poszczególnych metabolitów zostały skonstruowane przy użyciu stosunku powierzchni piku (powierzchnia piku metabolitu podzielona przez powierzchnię piku wybranego IS) każdego kalibratora do jego stężenia.
Wewnętrzne standardy składały się z 2H2NAM (dla NAM, methylNAM i ADPR) i 13C5;-Cyclic AMP (dla NMN i NAD+). Należy zauważyć, że ponieważ etykiety izotopowe nie są komercyjnie dostępne dla wszystkich metabolitów NAD, blisko spokrewniona cząsteczka (analog strukturalny) może być również użyta (Yamada i in., 2006), pod warunkiem, że jest uważana za podobną stabilność i wydajność jonizacji podczas analizy. Standardy wewnętrzne wybrane w tym badaniu zostały wcześniej zoptymalizowane dla powiązanego metabolitu (Bustamante et al., 2017).
Bezpieczeństwo
Bezpieczeństwo IV NAD+ oceniano za pomocą testów czynności wątroby i obserwacji klinicznej wszelkich zdarzeń niepożądanych. Testy czynnościowe wątroby obejmowały surowicę, bilirubinę całkowitą (bili), fosfatazę alkaliczną (ALP), aminotransferazę alaninową (ALT), gamma glutamylotransferazę (GGT), dehydrogenazę mleczanową (LD) i aminotransferazę asparaginianową (AST).
Analiza statystyczna
Analiza statystyczna została zakończona przy użyciu programu SPSS wersja 24 i GraphPad Prism wersja 8 dla okien. Do określenia, czy średnie stężenia dla badanych analitów różniły się w ciągu 8 h oraz między grupą badaną i kontrolną, zastosowano metodę średnich nieważonych, dwukierunkową ANOVA z testem wielokrotnych porównań Bonferroniego post hoc. Test Wilcoxona Signed Ranks został użyty do określenia, czy różnice w średnich stężeniach testów czynnościowych wątroby były istotne pomiędzy punktem czasowym linii podstawowej i 8 h. Różnice uznawano za istotne statystycznie, gdy p < 0,05.
Etyka
Badanie to przeprowadzono zgodnie z Kodeksem Etycznym Światowego Stowarzyszenia Medycznego (Deklaracja Helsińska) dla eksperymentów z udziałem ludzi. Zgodę etyczną uzyskano od William Carey University Institutional Review Board, Hattiesburg, MS (protokół #2017-12). Od wszystkich uczestników uzyskano świadomą zgodę.
Wyniki
Bezpieczeństwo
Nie zaobserwowano żadnych zdarzeń niepożądanych podczas 6-godzinnego wlewu z kohortami placebo (sól fizjologiczna) lub testowymi (NAD+).
Znaczące zmniejszenie aktywności (1,3, 57, 3,6 jednostek/L) dla enzymów czynnościowych wątroby, odpowiednio GGT, LD i AST, zaobserwowano po 8 h od rozpoczęcia wlewu NAD+ (Tabela 1). Żadna istotna zmiana aktywności dla jakiegokolwiek markera funkcji wątroby nie była widoczna w 8 h w próbkach otrzymujących placebo (sól fizjologiczną), jednakże niska liczba próbek może zmniejszać czułość dyskryminacji. Zaobserwowano również znaczący wzrost bilirubiny w osoczu o 2,75 μmola/L. Jednak żadna z tych zmian nie została uznana za istotną klinicznie.

Tabela 1. Test Wilcoxona Signed Ranks został użyty do określenia, czy różnice w średnich stężeniach testów czynnościowych wątroby były znaczące między punktami czasowymi linii podstawowej i 8 h.
Osocze
Ciągły wlew NAD+ z szybkością 3 μmoli/min spowodował znaczący (398%) wzrost poziomu NAD+ w osoczu tylko w punkcie czasowym 6 h (tj. koniec wlewu) w stosunku do linii podstawowej (p < 0,0001). Było to znacząco różne od kontroli 6 h traktowanej solą fizjologiczną (p < 0.001).
Poziomy NAD+ pozostały podwyższone w 8 h (tj, 2 h po infuzji) w stosunku do linii podstawowej i próbek kontrolnych traktowanych solą.
Poziomy NAD+ w osoczu nie zmieniły się znacząco od linii podstawowej w całym okresie oceny 8 h w próbkach kontrolnych traktowanych solą (Figura 1A).
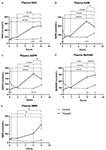
Rysunek 1. Zmiany w osoczu dinukleotydu nikotynamido-adeninowego (NAD+) i metabolitów w ciągu 8 h . (A) NAD+, (B) nikotynamid (NAM), (C) adenozynofosforybozy (ADPR), (D) metylo nikotynamid (meNAM), (E) mononukleotyd nikotynamidu (NMN). *p < 0,05, **p < 0,01, ***p < 0,001, ****p < 0,0001, jak wskazano. Dwukierunkowa ANOVA z testem wielokrotnego porównania Bonferroniego post hoc została użyta do określenia, czy średnie stężenia dla badanych analgetyków różniły się w okresie 8 h oraz pomiędzy grupami Test (n = 8) i Control (n = 3).
Podobnie do zmian obserwowanych dla NAD+, poziomy osocza metabolitu NAD+ nikotynamidu (NAM) wzrosły znacząco o 409% pod koniec infuzji NAD+ (tj, 6 h) w stosunku do linii podstawowej (p < 0,0001). Było to podobnie znacząco różne od kontroli 6 h traktowanej solą fizjologiczną (p < 0.001).
W punkcie czasowym 8 h (tj. 2 h po zakończeniu infuzji), poziomy NAM między grupą kontrolną i leczoną nie były już znacząco różne (p > 0.05).
Poziomy NAM dla próbek kontrolnych traktowanych solą fizjologiczną nie zmieniły się znacząco w okresie 8 h (p > 0.05, Figura 1B).
Zgodnie ze zmianami obserwowanymi dla NAM, poziomy osocza metabolitu NAD+ ADPR wzrosły znacząco o 393% na końcu infuzji NAD+ (tj. 6 h) w stosunku do linii podstawowej (p < 0,0001). Było to znacząco różne od kontroli traktowanej solą fizjologiczną 6 h (p < 0,0001).
W punkcie czasowym 8 h (tj. 2 h po zakończeniu infuzji) poziomy ADPR pozostały 305% powyżej linii podstawowej (p < 0,0001). Jednak nie było to znacząco większe niż poziomy ADPR w próbkach kontrolnych traktowanych solą fizjologiczną w tym samym punkcie czasowym (p > 0,05).
Poziomy ADPR dla próbek kontrolnych traktowanych solą fizjologiczną nie zmieniły się znacząco w całym 8-godzinnym okresie czasu (p > 0,05, Figura 1C).
Analiza korelacji Spearmana między średnimi grupowymi przez punkty czasowe 8 h dla dwóch metabolitów katabolicznych NAD+, NAM i ADPR, dała współczynnik korelacji 1,00 (p < 0.001).
Again, zgodnie ze zmianami obserwowanymi dla NAM, poziomy osocza metabolitu NAM, metylo-nikotynamidu (meNAM) wzrosły znacząco do 350% pod koniec infuzji NAD+ (tj, 6 h) w stosunku zarówno do linii podstawowej (p < 0,0001), jak i 6 h kontroli traktowanej solą fizjologiczną (p < 0,01).
W punkcie czasowym 8 h (tj, 2 h po zakończeniu infuzji) poziomy meNAM pozostawały 393% powyżej linii podstawowej (p < 0,0001) i znacznie większe niż próbki kontrolne traktowane solą fizjologiczną w tym samym punkcie czasowym (p < 0,05).
PoziomymeNAM dla próbek kontrolnych traktowanych solą fizjologiczną nie zmieniły się znacząco w całym 8-godzinnym okresie czasu (p > 0,05, Figura 1D).
Poziomy NMN, metabolit NAM poprzez anaboliczną ścieżkę zbawczą, były znacząco podwyższone (472%) tylko w 8-godzinnym punkcie czasowym (tj, 2 h po zakończeniu infuzji, p < 0,05).
NMN poziomy dla próbek kontrolnych traktowanych solą fizjologiczną nie zmieniły się znacząco w całym 8-godzinnym okresie czasu (p > 0,05, Figura 1E).
Mocz
Ciągły dożylny wlew NAD+ z szybkością 3 μmoli/min spowodował znaczący (538%) wzrost szybkości wydalania NAD+ z moczem w punkcie czasowym 6 h (tj, koniec infuzji) w stosunku do wydalanego w 30 min (p < 0,001). Było to również znacząco różne od ilości NAD+ wydalanego z moczem w 6 h przez kontrole traktowane solą fizjologiczną (p < 0,05).
Ilość NAD+ wydalanego w moczu zmniejszyła się o 43% (p < 0,05) w 8 h (tj, 2 h po infuzji) w stosunku do szczytowego wydalania w punkcie czasowym 6 h.
Szybkość wydalania NAD+ z moczem nie zmieniła się znacząco w okresie oceny 8 h w próbkach kontrolnych traktowanych solą fizjologiczną (Figura 2A).

Rysunek 2. Zmiany w moczu NAD+ i metabolitów w ciągu 8 h (pierwsze 6 h składające się z ciągłego, 3 μmoli/min, wlewu dożylnego NAD+). (A) NAD+, (B) NAM, (C) meNAM. *p < 0,05, **p < 0,01, ***p < 0,001, ****p < 0,0001, jak wskazano. Dwukierunkowa ANOVA z testem wielokrotnego porównania Bonferroniego post hoc została użyta do określenia, czy średnie stężenia dla badanych analitów były różne w okresie 8 h oraz pomiędzy grupami Test (n = 8) i Control (n = 3).
Szybkość wydalania metabolitu NAD+ NAM nie zmieniała się znacząco w całym okresie 8 h badania i nie różniła się od obserwowanej szybkości wydalania NAM dla kontroli traktowanych solą fizjologiczną (Figura 2B).
Szybkość wydalania z moczem metabolitu NAM meNAM była znacząco zwiększona (403%) w punkcie czasowym 6 h (tj, koniec infuzji) w stosunku do obserwowanego w 30 min (p < 0,01). Ilość meNAM wydalana z moczem zmniejszyła się o 43% (p < 0,05) w 8 h (tj, 2 h po infuzji) w stosunku do szczytowego wydalania w punkcie czasowym 6 h.
Szybkość wydalania meNAM z moczem nie zmieniła się znacząco w okresie oceny 8 h w kontrolach traktowanych solą fizjologiczną (Figura 2C).
Dyskusja
Rosnące zainteresowanie terapiami opartymi na NAD+, w tym infuzjami NAD+, uwydatniło potrzebę wyraźniejszego zrozumienia losu NAD+ i jego metabolitów po podaniu dożylnym. Obecne badanie dokumentuje zmiany w poziomach NAD+ i kluczowych metabolitów zarówno w osoczu, jak i w moczu w ciągu 8 godzin przy zastosowaniu typowego klinicznego schematu dawkowania 750 mg NAD+ podawanego dożylnie w ciągu 6 godzin.
Co ważne, infuzja NAD+ nie spowodowała żadnych obserwowalnych zdarzeń niepożądanych w badanej kohorcie, a raczej zmniejszyła aktywność osoczową enzymów wskazujących na stres wątrobowy, takich jak wewnątrzwątrobowa LD i AST oraz enzymu po-wątrobowego (przewód żółciowy) GGT, sugerując, że integralność zarówno wewnątrzwątrobowej, jak i po-wątrobowej tkanki została wzmocniona nawet w stosunkowo krótkich ramach czasowych 8 h. Wzrost bilirubiny, produktu degradacji krwinek czerwonych, w 8 godzinie może odzwierciedlać albo bardzo mały wzrost obrotu krwinek czerwonych, co może wystąpić w wyniku hemolizy indukowanej infuzją, albo zmniejszony metabolizm hemu (Tabela 1). Jednakże, biorąc pod uwagę bardzo małą wielkość tej zmiany, nie uznano tego za istotne klinicznie.
Jak oczekiwano, u uczestników leczonych solą (tj. kontrola), poziomy osocza NAD+ i metabolitów NAM i ADPR oraz metabolitów NAM, NMN i meNAM pozostały zasadniczo niezmienione przez okres 8 h. Jednakże między 30 min a 6 h zaobserwowano wyraźny spadek NAM, meNAM i ADPR, najprawdopodobniej spowodowany efektem rozcieńczenia solą fizjologiczną. Zgodnie z tą koncepcją, wartości dla tych analitów powróciły do poziomu wyjściowego w 8 h (tj. 2 h po zakończeniu podawania soli), 2 h po zakończeniu infuzji soli).
Niespodziewanie jednak u uczestników infuzji NAD+, poziomy NAD+ w osoczu nie wzrosły do czasu po punkcie czasowym 2 h, osiągając maksimum ~400% powyżej linii podstawowej dla NAD+ i metabolitów NAM, meNAM i ADPR (odpowiednio 398%, 409%, 393%) tylko w punkcie czasowym 6 h (Figury 1A-E). Było to wewnętrznie spójne ze szczytem wydalania z moczem zarówno NAD+ jak i meNAM również występującym w 6 h przed gwałtownym spadkiem po zakończeniu infuzji (ryc. 2A-C).
NAD+ był infuzowany ze stałą szybkością 3 μmoli/min. Dlatego 90 μmoli NAD+ było infuzowane bezpośrednio do przedziału naczyniowego co 30 min dostarczając łącznie 1080 μmoli do końca infuzji w 6 h. Mieszanie wewnątrznaczyniowe z dowolnego punktu infuzji zachodzi w ciągu ~2 min; zakładając średnią objętość krwi 5, dodany NAD+, przy braku znaczącego metabolizmu lub absorpcji, odpowiadałby dodatkowemu wzrostowi (NAD+) o co najmniej 18 μM, co 30 min przez 6 h infuzji. Podczas gdy wzrost tej wielkości mieści się w granicach wykrywalności analitycznej tego badania, nie zaobserwowano wzrostu NAD+ ani jego metabolitów aż do 2 h (tj. w punkcie czasowym 6 h) ani w osoczu ani w moczu. Ta nieoczekiwana obserwacja wskazuje na szybki, i co najmniej przez pierwsze 2 h, całkowity wychwyt tkankowy i/lub metabolizm NAD+ i/lub jego metabolitów.
Liczba enzymów może osiągnąć skuteczny katabolizm NAD+, w tym sirtuiny (SIRTs 1-7) transferazy adenozynodifosforanu (ADP)-rybozy (ARTs) i polimerazy poli(ADP-rybozy) (PARPs 1-17) oraz syntazy cyklicznej ADP-rybozy (cADPR) (CD38, CD157). Pozakomórkowe pirofosfatazy NAD+ obecne w ludzkiej surowicy mogą również degradować NAD+ do AMP i NMN (Schmidt-Brauns i in., 2001). Białko powierzchniowe komórki CD73 również przekształca NMN w NR, który łatwo przekracza błony komórkowe w celu potencjalnej resyntezy do NAD+. Co ważne, glikohydrolazy katabolizujące NAD+, CD38 i CD157, pirofosfataza ektonukleotydowa (CD203a) i katabolizator NMN CD73 są ektoenzymami występującymi na wielu różnych komórkach, w tym limfoidalnych, granulocytarnych, neuronalnych i śródbłonkowych (Wei i in., 2014), a rozpuszczalne w osoczu glikohydrolazy NAD mogą być również obecne (rysunek 3; Funaro i in., 2009). Równoległy wzrost NAM i ADPR w osoczu (współczynnik korelacji 1,000, p < 0,001, dane nie pokazane) silnie sugeruje, że przynajmniej do 6 h głównym przeznaczeniem NAD + jest metabolizm poprzez rozszczepienie glikozydowego wiązania ADPribozy-nikotynamidowego do NAM i ADPR, produktów ubocznych symptomatycznych dla aktywności glikohydrolazy NAD (np. CD38). Jest to zgodne z dowodami innych, gdzie CD38, w szczególności, wykazano, że ma podstawową rolę w kontrolowaniu pozakomórkowych poziomów NAD+ (Wei i in., 2014). Dorosłe ludzkie erytrocyty są CD38 pozytywne i wyrażają wysoki poziom aktywności glikohydrolazy NAD+, rozszczepiając egzogenny NAD+ w celu dostarczenia erytrocytom rybozy ADP, która może być skutecznie pobrana do komórki (Kim i in., 1993; Albeniz i in., 2004). Brak jakiegokolwiek wzrostu NAD+ lub metabolitów, w osoczu lub moczu, do czasu po pierwszych 2 h infuzji wskazuje, że NAD+ i/lub jego metabolity są przemieszczane z zewnątrzkomórkowej przestrzeni naczyniowej i skutecznie sekwestrowane w tkankach lub przedziałach pozanaczyniowych jako NAD+ i/lub jego metabolity w tym okresie czasu.

Rysunek 3. Potencjalny zewnątrzkomórkowy katabolizm egzogennie dostarczonego NAD+ poprzez aktywność ektoenzymów CD38 (syntaza ADP-rybozy (ADPR)), CD203a (pirofosfataza NAD+), CD 73 (5′-nukleotydaza), CD157-ADP-rybozylowa cyklaza 2. Skróty: NAM, nikotynamid; NMN, mononukleotyd nikotynamidu; ADPR, rybozyd adenozynodifosforanu; meNAM, nikotynamid metylu; 4PY, N-metylo-4-pirydono-3-karboksyamid; 2PY, metylo-2-pirydono-5-karboksyamid; ARPP, pirofosfataza rybozy ADP; RPPK, pirofosfokinaza rybozylu; NAmpt, fosforybozylotransferaza nikotynamidowa; NMNAT, mononukleotydadenylotransferaza nikotynamidowa; CX-43, connexin 43.
Chociaż PARP i sirtuiny są zaangażowane w katabolizm NAD+, jako enzymy wewnątrzkomórkowe z przedstawicielami w cytoplazmie, jądrze i mitochondriach raczej nie wpływają bezpośrednio na pozakomórkowe poziomy NAD+. Można jednak oczekiwać, że enzymy te będą reagować na zmiany wewnątrzkomórkowych stężeń NAD+, NAM i NR, które mogą powstać w wyniku egzogennie dostarczonego NAD+. Rysunek 4 podsumowuje schematycznie możliwe losy egzogennego NAD+.
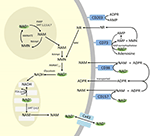
Rysunek 4. Potencjalne wewnątrz- i zewnątrzkomórkowe losy egzogennie dostarczanego NAD+. Skróty: CD38 , CD203a (pirofosfataza NAD+), CD 73 (5′-nukleotydaza). NAM, nikotynamid; NMN, mononukleotyd nikotynamidu; ADPR, rybozyd adenozynodifosforanu; meNAM, nikotynamid metylu; 4PY, N-metylo-4-pirydono-3-karboksyamid; 2PY, metylo-2-pirydono-5-karboksyamid; ARPP, pirofosfataza rybozy ADP; RPPK, pirofosfokinaza rybozylu; NAmpt, fosforybozylotransferaza nikotynamidowa; NMNAT, mononukleotydadenylotransferaza nikotynamidowa; PARP ; NRK .
Podczas gdy istnieje znaczna zdolność do szybkiej degradacji egzogennie dostarczanego IV NAD+ do metabolitów składowych, warto również zauważyć, że może również występować wychwyt pozakomórkowego NAD+. Ponieważ NAD+, ma ogólny ładunek ujemny, nie jest w stanie biernie przekraczać błon komórkowych i dlatego musi być aktywnie transportowany przez membranę. To, że tak się dzieje, zostało wykazane przez wielu badaczy, którzy donieśli, że egzogenny NAD+ aplikowany do różnych typów ludzkich komórek rzeczywiście powoduje znaczne podwyższenie wewnątrzkomórkowego NAD+ (Ying et al., 2003; Zhu et al., 2005; Billington et al., 2008; Pittelli et al., 2011; Felici et al., 2013). Chociaż zaangażowany mechanizm(y) nie został jeszcze w pełni scharakteryzowany, Alano i wsp. (2010) donoszą, że egzogenny NAD+ może wnikać do neuronów przez kanały bramkowane P2X7, a inni konsekwentnie obserwują transport NAD+ przez błony za pośrednictwem hemichannels connexin 43 (CX43), nawet w stężeniach tak niskich jak 250 pM (Billington i wsp., 2008). Ponieważ koneksyny są szeroko rozpowszechnione w tkankach ludzkich, a CX43 wydaje się być najbardziej wszechobecną koneksyną w wielu typach komórek, nie można wykluczyć możliwości szybkiego wychwytu NAD+.
Więc komórkowy pobór i/lub metabolizm NAD+ i metabolitów NAM i ADPR i wtórnych metabolitów meNAM i NMN wydawał się postępować w tempie z 3 μmoles/min NAD+ infuzji dla przynajmniej pierwszych 2 h. Albo NAD+ i / lub pierwotne badane metabolity są skutecznie sekwestrowane podczas tego pierwszego 2-6 h okresu lub wtórne metabolity są również tworzone. ADPR może być poddawany recyklingowi w celu wytworzenia NAD+ z NAM (Figura 4) lub dalej metabolizowany przez ektoenzymy takie jak CD203a (pirofosfataza NAD+) w celu wytworzenia AMP, który może być dalej szybko metabolizowany do adenozyny przez CD73 a 5′-nukleotydazę (Bogan i Brenner, 2010; Horenstein i in., 2016; Morandi i in., 2018). Na poparcie tej hipotezy, wzrost poziomu adenozyny we krwi został uznany przez innych jako konsekwencja infuzji pozakomórkowego NAD+ (Szczepańska-Konkel i in., 2003). NAM może być również metabolizowany do NMN na drodze salwatoryjnej przed resyntezą do NAD+ (ryc. 3, 4) lub pod wpływem metylotransferaz wątrobowych wytwarzany jest N1-metylonikotynamid, który może być wydalany bezpośrednio lub dalej przekształcany do N-metylo-2-pirydono-5-karboksyamidu (2PY, +99% meNAM) i N-metylo-4-pirydono-3-karboksyamidu (4PY, ~0.25% meNAM) przed wydaleniem (Shibata i Matsuo, 1989; Okamoto i in…, 2003).
Jest oczywiste, że przy stosowanym reżimie dawkowania mechanizmy zaangażowane w metabolizm i sekwestrację NAD+ i metabolitów osiągnęły nasycenie gdzieś po 2 h, co spowodowało znaczne podwyższenie NAD+ w osoczu i akumulację wszystkich badanych metabolitów (NMA, ADPR, meNAM i NMN) w 6 h. Jak wspomniano wcześniej dane te wspierają pogląd, że jedną z głównych dróg metabolizmu NAD+ w tych warunkach jest rozszczepienie wiązania glikozydowego, przez ektoenzymy takie jak CD38 do produkcji NAM i ADPR. Jednak wzrost NMN w osoczu po 2 h sugeruje również, że egzogenny NAD+ jest prawdopodobnie działany przez zewnątrzkomórkowe pirofosfatazy NAD+, podnosząc NMN w osoczu i uwalniając AMP jako dodatkowy metabolit.
Podsumowując, to badanie ujawniło po raz pierwszy, że: (a) przy szybkości przepływu 3 μmole/min cały egzogennie infuzowany NAD+ był szybko i całkowicie usuwany z osocza przez co najmniej pierwsze 2 h; (b) analizowany wzrost bioproduktów metabolicznych jest zgodny z aktywnością glikohydrolaz NAD+ i pirofosfatazy NAD+; oraz (c) produkty wydalania z moczem powstające w wyniku infuzji NAD+ obejmują natywny NAD+ i meNAM, ale nie NAM.
Pomimo, że ustalenia tego badania są nowatorskie i zmierzają w pewnym stopniu do pogłębienia naszego zrozumienia czasowego losu egzogennego NAD+ u ludzi, zidentyfikowano ograniczenia. Aby poprawić ogólne metaboliczne przeliczanie przyszłe badania powinny zbadać zmiany w czerwonych komórkach NAD+ i metabolitów i wydalanie z moczem wtórnych metabolitów meNAM, 2PY i 4PY. Ponadto, prawdopodobnie przydatna będzie ocena wpływu egzogennego NAD+ na zmiany w metabolizmie puryn, która powinna obejmować co najmniej ocenę osoczowego i czerwonokrwinkowego AMP i adenozyny. Zastosowanie modeli zwierzęcych może również pomóc w wyjaśnieniu względnego zaangażowania różnych szlaków metabolicznych poprzez zastosowanie odpowiednich inhibitorów farmakologicznych i pobieranie próbek tkanek.
W podsumowaniu, to badanie było w stanie ujawnić po raz pierwszy kilka bardzo użytecznych, wcześniej nieznanych, informacji o losie egzogennego IV NAD+ u ludzi, w tym, ogólne bezpieczeństwo i tolerancja infuzji IV NAD+ w tempie 3 μmoli/min, szybkiego sekwestrowania NAD+ z osocza, prawdopodobnego udziału zarówno glikohydrolazy NAD+ jak i aktywności pirofosforanu NAD+ w metabolizmie NAD+ oraz najwyraźniej skutecznej reabsorpcji NAM w kanalikach nerkowych. Konieczne jest jednak przeprowadzenie dodatkowych badań w celu pełnego ujawnienia złożonego metabolicznego losu tej ważnej cząsteczki. Ponadto, charakterystyka tych zmian pomoże w rozwoju i poprawie schematów leczenia opartych na NAD+ dla zaburzeń, które prawdopodobnie skorzystają ze zwiększonej dostępności NAD+, w tym warunków wymagających zwiększonej regeneracji i naprawy komórkowej, takich jak choroba Alzheimera i inne demencje neurodegeneracyjne.
Dostępność danych
Zestawy danych wygenerowane dla tego badania są dostępne na żądanie do odpowiedniego autora.
Oświadczenie o etyce
Badania z udziałem ludzi zostały przejrzane i zatwierdzone przez William Carey University Institutional Review Board, Hattiesburg, MS (protokół #2017-12). Pacjenci/uczestnicy wyrazili pisemną świadomą zgodę na udział w tym badaniu.
Wkład autorów
RG uczestniczył w projektowaniu i nadzorowaniu badania, krytycznej dyskusji i autorstwie manuskryptu. JBer uczestniczył w projektowaniu badania, krytycznej dyskusji, zbieraniu danych, analizie statystycznej, redagowaniu i przeglądzie manuskryptu. RM uczestniczył w projektowaniu badania, krytycznej dyskusji, nadzorze klinicznym i przeglądzie manuskryptu. NB brał udział w analizie biochemicznej i przeglądzie manuskryptu. JW brał udział w projektowaniu badania, krytycznej dyskusji i przeglądzie manuskryptu. JBen uczestniczył w zbieraniu danych i przeglądzie manuskryptu. SB uczestniczył w projektowaniu badania i przeglądzie manuskryptu. Wszyscy autorzy przejrzeli, przeczytali i zatwierdzili ostateczną wersję manuskryptu i zgadzają się z kolejnością prezentacji autorów.
Funding
This study was funded jointly by the NAD+ Research Inc, (LA, USA) oraz Australasian Research Institute Incorporation (Sydney, Australia) i zostało przeprowadzone w Springfield Wellness Center, Springfield, LA, USA.
Oświadczenie o konflikcie interesów
RM jest dyrektorem NAD+ Research Inc. oraz dyrektorem medycznym Springfield Wellness Center, które stosuje IV NAD jako terapię kliniczną. SB otrzymał honoraria za konsultacje od NAD+ Research Inc.
Pozostali autorzy oświadczają, że badania zostały przeprowadzone przy braku jakichkolwiek komercyjnych lub finansowych powiązań, które mogłyby być interpretowane jako potencjalny konflikt interesów.
Podziękowania
Pragniemy podziękować Archway Apothecary Pty Limited, LA, USA za nieodpłatne dostarczenie IV NAD+ na potrzeby tego projektu.
Abeti, R., and Duchen, M. R. (2012). Activation of PARP by oxidative stress induced by β-amyloid: implications for Alzheimer’s disease. Neurochem. Res. 37, 2589-2596. doi: 10.1007/s11064-012-0895-x
PubMed Abstract | CrossRef Full Text | Google Scholar
Airhart, S. E., Shireman, L. M., Risler, L. J., Anderson, G. D., Nagana Gowda, G. A., Raftery, D., et al. (2017). An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS One 12:e0186459. doi: 10.1371/journal.pone.0186459
PubMed Abstract | CrossRef Full Text | Google Scholar
Alano, C., Garnier, P., Ying, W., Higashi, Y., Kauppinen, T. M., and Swanson, R. A. (2010). NAD+ depletion is necessary and sufficient for PARP-1 mediated neuronal death. J. Neurosci. 30, 2967-2978. doi: 10.1523/JNEUROSCI.5552-09.2010
PubMed Abstract | CrossRef Full Text | Google Scholar
Albeniz, I., Demir, O., Nurten, R., and Bermek, E. (2004). NAD glycohydrolase activities and adp-ribose uptake in erythrocytes from normal subjects and cancer patients. Biosci. Rep. 24, 41-53. doi: 10.1023/b:bire.0000037755.42767.a4
PubMed Abstract | CrossRef Full Text | Google Scholar
Billington, R. A., Travelli, C., Ercolano, E., Galli, U., Roman, C. B., Grolla, A. A., et al. (2008). Characterisation of NAD+ uptake in mammalian cells. J. Biol. Chem. 283, 6367-6374. doi: 10.1074/jbc.M706204200
PubMed Abstract | CrossRef Full Text | Google Scholar
Bogan, K., and Brenner, C. (2010). 5′-nukleotydazy i ich nowe role w metabolizmie NAD+ i fosforanów. New J. Chem. 34, 845-853. doi: 10.1039/b9nj00758j
CrossRef Full Text | Google Scholar
Braidy, N., Guillemin, G. J., Mansour, H., Chan-Ling, T., Poljak, A., and Grant, R. (2011). Age related changes in NAD+ metabolism oxidative stress and sirt1 activity in wistar rats. PLoS One 6:e19194. doi: 10.1371/journal.pone.0019194
PubMed Abstract | CrossRef Full Text | Google Scholar
Braidy, N., Poljak, A., Grant, R., Jayasena, T., Mansour, H., Chan-Ling, T., et al. (2014). Mapping NAD+ metabolism in the brain of ageing Wistar rats: potential targets for influencing brain senescence. Biogerontology 15, 177-198. doi: 10.1007/s10522-013-9489-5
PubMed Abstract | CrossRef Full Text | Google Scholar
Bustamante, S., Jayasena, T., Richani, D., Gilchrist, R., Wu, L., Sinclair, D., et al. (2017). Quantifying the cellular NAD+ metabolome using a tandem liquid chromatography mass spectrometry approach. Metabolomics 14:15. doi: 10.1007/s11306-017-1310-z
PubMed Abstract | CrossRef Full Text | Google Scholar
Clement, J., Wong, M., Poljak, A., Sachdev, P., and Braidy, N. (2018). Metabolom NAD+ osocza jest dysregulowany w „normalnym” starzeniu się. Rejuvenation Res. 22, 121-130. doi: 10.1089/rej.2018.2077
PubMed Abstract | CrossRef Full Text | Google Scholar
Csiszar, A., Tarantini, S., Yabluchanskiy, A., Balasubramanian, P., Kiss, T., Farkas, E., et al. (2019). Role of endothelial NAD+ deficiency in age-related vascular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 316, H1253-H1266. doi: 10.1152/ajpheart.00039.2019
PubMed Abstract | CrossRef Full Text | Google Scholar
Essuman, K., Summers, D. W., Sasaki, Y., Mao, X., DiAntonio, A., and Milbrandt, J. (2017). The SARM1 toll/interleukin-1 receptor domain possesses intrinsic NAD+ cleavage activity that promotes pathological axonal degeneration. Neuron 93, 1334.e5-1343.e5. doi: 10.1016/j.neuron.2017.02.022
PubMed Abstract | CrossRef Full Text | Google Scholar
Felici, R., Lapucci, A., Ramazzotti, M., and Chiarugi, A. (2013). Insight into molecular and functional properties of NMNAT3 reveals new hints of NAD homeostasis within human mitochondria. PLoS One 8:e76938. doi: 10.1371/journal.pone.0076938
PubMed Abstract | CrossRef Full Text | Google Scholar
Funaro, A., Ortolan, E., Bovino, P., Lo Buono, N., Nacci, G., Parrotta, R., et al. (2009). Ectoenzymes and innate immunity: the role of human CD157 in leukocyte trafficking. Front. Biosci. 14, 929-943. doi: 10.2741/3287
PubMed Abstract | CrossRef Full Text | Google Scholar
Goldberger, J. (1914). Etiologia Pellagry: znaczenie niektórych obserwacji epidemiologicznych w odniesieniu do niej. Public Health Rep. 29, 1683-1686. doi: 10.2307/4570920
CrossRef Full Text | Google Scholar
Guest, J., Grant, R., Mori, T. A., and Croft, K. D. (2014). Zmiany w uszkodzeniach oksydacyjnych, stanach zapalnych i z wiekiem w płynie mózgowo-rdzeniowym. PLoS One 9:e85335. doi: 10.1371/journal.pone.0085335
PubMed Abstract | CrossRef Full Text | Google Scholar
Horenstein, A., Quarona, V., Toscani, D., Costa, F., Chillemi, A., Pistoia, V., et al. (2016). Adenozyna generowana w niszy szpiku kostnego poprzez szlak CD38-mediated koreluje z progresją szpiczaka ludzkiego. Mol. Med. 22, 694-704. doi: 10.2119/molmed.2016.00198
PubMed Abstract | CrossRef Full Text | Google Scholar
Kim, U., Han, M. K., Park, B. H., Kim, H. R., and An, N. H. (1993). Functionof NAD glycohydorlase in ADP-ribose uptake from human erythrocytes. Biochim. Biophys. Acta 1178, 121-126. doi: 10.1016/0167-4889(93)90001-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Long, A. N., Owens, K., Schlappal, A. E., Kristian, T., Fishman, P. S., and Schuh, R. A. (2015). Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’ disease-relevant murine model. BMC Neurol. 15:19. doi: 10.1186/s12883-015-0272-x
PubMed Abstract | CrossRef Full Text | Google Scholar
Massudi, H., Grant, R., Braidy, N., Guest, J., Farnsworth, B., Guillemin, G. J., et al. (2012). Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 7:e42357. doi: 10.1371/journal.pone.0042357
PubMed Abstract | CrossRef Full Text | Google Scholar
Mestayer, P. N. (2019). Uzależnienie ciemna noc duszy, NAD+ światło nadziei. (Bloomington IS: Balboa Press).
Google Scholar
Mills, K. F., Yoshida, S., Stein, L. R., Grozio, A., Kubota, S., Sasaki, Y., et al. (2016). Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metab. 24, 795-806. doi: 10.1016/j.cmet.2016.09.013
PubMed Abstract | CrossRef Full Text | Google Scholar
Morandi, F., Horenstein, A. L., Rizzo, R., and Malavasi, F. (2018). Rola zewnątrzkomórkowej generacji adenozyny w rozwoju chorób autoimmunologicznych. Mediators Inflamm. 2018:7019398. doi: 10.1155/2018/7019398
PubMed Abstract | CrossRef Full Text | Google Scholar
O’Holleran, P. (1961). DPN w zapobieganiu, diagnozowaniu i leczeniu uzależnień od narkotyków. West. J. Surg. Obst. Gyn. 69, 213-215.
Google Scholar
Okamoto, H., Ishikawa, A., Yoshitake, Y., Kodama, N., Nishimuta, M., Fukuwatari, T., et al. (2003). Diurnal variations in human urinary excretion of nicotinamide catabolites: effects of stress on the metabolism of nicotinamide. Am. J. Clin. Nutr. 77, 406-410. doi: 10.1093/ajcn/77.2.406
PubMed Abstract | CrossRef Full Text | Google Scholar
Pillai, J. B., Isbatan, A., Imai, S., and Gupta, M. P. (2005). Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2α deacetylase activity. J. Biol. Chem. 280, 43121-43130. doi: 10.1074/jbc.m506162200
PubMed Abstract | CrossRef Full Text | Google Scholar
Pittelli, M., Felici, R., Pitozzi, V., Giovannelli, L., Bigagli, E., Cialdai, F., et al. (2011). Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol. Pharmacol. 80, 1136-1146. doi: 10.1124/mol.111.073916
PubMed Abstract | CrossRef Full Text | Google Scholar
Schmidt-Brauns, J., Herbert, M., Kemmer, G., Kraiss, A., Schlör, S., and Reidl, J. (2001). Is a NAD pyrophosphatase activity necessary for Haemophilus influenzae type b multiplication in the blood stream? Int. J. Med. Microbiol. 291, 219-225. doi: 10.1078/1438-4221-00122
PubMed Abstract | CrossRef Full Text | Google Scholar
Shibata, K., and Matsuo, H. (1989). Korelacja między spożyciem równoważnika niacyny i wydalaniem z moczem jej metabolitów, N’-metylonikotynamidu, N’-metylo-2-pirydono-5-karboksyamidu i N’-metylo-4-pirydono-3-karboksyamidu, u ludzi spożywających samodzielnie wybraną żywność. Am. J. Clin. Nutr. 50, 114-119. doi: 10.1093/ajcn/50.1.114
PubMed Abstract | CrossRef Full Text | Google Scholar
Su, B., Wang, X., Nunomura, A., Moreira, P., Lee, H. G., Perry, G., et al. (2008). Oxidative stress signaling in Alzheimer’s disease. Curr. Alzheimer Res. 5, 525-532.
Google Scholar
Szczepańska-Konkel, M., Langner, G., Bednarczuk, G., Stiepanow-Trzeciak, A., Jankowski, M., and Angielski, S. (2003). Renal hemodynamics and natriuretic responses to intravenous administration of diadenosine tetraphosphate (ap4a) and nicotinamide adenine dinucleotide (NAD) in rat. J. Physiol. Pharmacol. 54, 163-173.
PubMed Abstract | Google Scholar
Wei, W., Graeff, R., and Yue, J. (2014). Roles and mechanisms of the CD38/cyclic adenosine diphosphate ribose/Ca2+ signaling pathway. World J. Biol. Chem. 5, 58-67. doi: 10.4331/wjbc.v5.i1.58
PubMed Abstract | CrossRef Full Text | Google Scholar
Wu, J., Jin, Z., Zheng, H., and Yan, L. J. (2016). Sources and implications of NADH/NAD+ redox imbalance in diabetes and its complications. Diabetes Metab. Syndr. Obes. 9, 145-153. doi: 10.2147/dmso.s106087
PubMed Abstract | CrossRef Full Text | Google Scholar
Yamada, K., Hara, N., Shibata, T., Osago, H., and Tsuchiya, M. (2006). The simultaneous measurement of nicotinamide adenine dinucleotide and related compounds by liquid chromatography/electrospray ionization tandem mass spectrometry. Anal. Biochem. 352, 282-285. doi: 10.1016/j.ab.2006.02.017
PubMed Abstract | CrossRef Full Text | Google Scholar
Ying, W. (2013). Roles of NAD+, PARP-1, and sirtuins in cell death, ischemic brain injury, and synchrotron radiation X-ray-induced tissue injury. Scientifica 2013:691251. doi: 10.1155/2013/691251
PubMed Abstract | CrossRef Full Text | Google Scholar
Ying, W., Garnier, P., and Swanson, R. A. (2003). NAD+ repletion prevents PARP-1-induced glycolytic blockade and cell death in cultured mouse astrocytes. Biochem. Biophys. Res. Commun. 308, 809-813. doi: 10.1016/s0006-291x(03)01483-9
PubMed Abstract | CrossRef Full Text | Google Scholar
Ying, W., and Xiong, Z.-G. (2010). Oxidative stress and NAD+ in ischemic brain injury: current advances and future perspectives. Curr. Med. Chem. 17, 2152-2158. doi: 10.2174/092986710791299911
PubMed Abstract | CrossRef Full Text | Google Scholar
Yoshino, J., Baur, J. A., and Imai, S. I. (2018). Półprodukty NAD+: biologia i potencjał terapeutyczny NMN i NR. Cell Metab. 27, 513-528. doi: 10.1016/j.cmet.2017.11.002
PubMed Abstract | CrossRef Full Text | Google Scholar
Yoshino, J., Mills, K. F., Yoon, M. J., and Imai, S. (2011). Mononukleotyd nikotynamidowy, kluczowy pośrednik NAD+, leczy patofizjologię cukrzycy wywołanej dietą i wiekiem u myszy. Cell Metab. 14, 528-536. doi: 10.1016/j.cmet.2011.08.014
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhou, M., Ottenberg, G., Sferrazza, G. F., Hubbs, C., Fallahi, M., Rumbaugh, G., et al. (2015). Neuronal death induced by misfolded prion protein is due to NAD+ depletion and can be relieved in vitro and in vivo by NAD+ replenishment. Brain 138, 992-1008. doi: 10.1093/brain/awv002
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhu, K., Swanson, R. A., and Ying, W. (2005). NADH może wejść do astrocytów, aby zablokować PARP-1-mediowaną śmierć astrocytów. Neuroreport 16, 1209-1212. doi: 10.1097/00001756-200508010-00015
PubMed Abstract | CrossRef Full Text | Google Scholar
Zhua, X., Sua, B., Wanga, X., Smitha, M. A., and Perry, G. (2007). Causes of oxidative stress in Alzheimer disease. Cell. Mol. Life Sci. 64, 2202-2210. doi: 10.1007/s00018-007-7218-4
PubMed Abstract | CrossRef Full Text | Google Scholar
.