I segnali intracellulari, come i danni al DNA della cellula, guidano l’apoptosi principalmente attraverso il percorso intrinseco. La via intrinseca dell’apoptosi, che coinvolge proteine di segnalazione conservate, è fisicamente associata ai mitocondri, e nei vertebrati è sensibile allo stress ossidativo mitocondriale. La via è influenzata dai membri della famiglia Bcl legati alla membrana mitocondriale, tra cui il gene Bax e Bcl-2, che agiscono come proteine regolatrici pro- o anti-apoptotiche, rispettivamente.
Panoramica della via dell’apoptosi intrinseca
La via dell’apoptosi intrinseca è iniziata, per esempio, dalla chemioterapia e/o radioterapia. È attivata da una serie di stimoli esogeni ed endogeni, come il danno al DNA, l’ischemia e lo stress ossidativo. Inoltre, svolge una funzione importante nello sviluppo e nell’eliminazione delle cellule danneggiate.
Nella via intrinseca, la conseguenza funzionale della segnalazione pro-apoptotica è la perturbazione della membrana mitocondriale e il rilascio del citocromo c nel citoplasma, dove forma un complesso o apoptosoma con il fattore attivante la proteasi apoptotica 1 (APAF1) e la forma inattiva della caspasi-9. Questo complesso idrolizza l’adenosina trifosfato per scindere e attivare la caspasi-9. L’iniziatore caspasi-9 poi scinde e attiva le caspasi esecutrici-3/6/7, provocando l’apoptosi cellulare. È totalmente diverso dai segnali extracellulari, che sono di solito generati dalle cellule citotossiche del sistema immunitario e innescano l’apoptosi principalmente attraverso la via estrinseca.
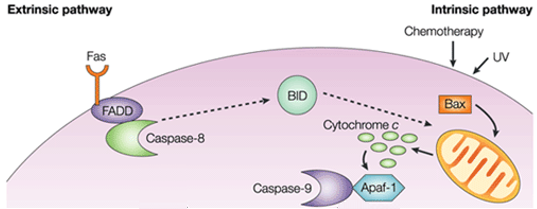
Figura 1. Differenza tra la via estrinseca e la via intrinseca.
Processo e regolazione della via dell’apoptosi intrinseca
La via dell’apoptosi intrinseca induce l’apoptosi attivando direttamente la caspasi-3 o scindendo il bid (BH3 interacting domain death agonist), con conseguente disfunzione mitocondriale e successivo rilascio di citocromo c e attivazione delle caspasi-9 e caspasi-3. La caspasi-3 promuove le caratteristiche tipiche dell’apoptosi, tra cui la frammentazione del DNA e la morte cellulare in diversi tessuti.
La famiglia di proteine B-cell lymphoma 2 (Bcl-2) controlla strettamente l’attivazione della via intrinseca. Si trova nel linfoma follicolare ed è stata identificata per la prima volta come uno dei geni coinvolti nella morte cellulare attivando l’apoptosi pro-apoptotica o inibendo quella anti-apoptotica. Le proteine di un sottogruppo, tra cui Bid, Bad, Bim, Bmf, Puma e Noxa, contengono un singolo dominio Bcl-2 homology 3 (proteine solo BH3) e hanno attività pro-apoptotica. Altri due sottoinsiemi di proteine hanno domini BH multipli. Il primo sottoinsieme, che include la proteina X associata a Bcl-2 (Bax), l’antagonista/killer omologo di Bcl-2 (Bak) e il regolatore di apoptosi della famiglia Bcl-2 (Bok), è pro-apoptotico; l’altro sottoinsieme, che include Bcl-2, Bcl-XL e Mcl-1, è anti-apoptotico. Il percorso mitocondriale è in parte influenzato dai membri della famiglia Bcl legati alla membrana mitocondriale, comprese le proteine regolatrici pro-apoptotiche Bax e quelle anti-apoptotiche Bcl-2.
Le molecole pro-apoptotiche causano la permeabilizzazione della membrana mitocondriale esterna, portando all’efflusso del citocromo c, che lega l’adattatore Apaf-1 e l’iniziatore caspasi-9 nel citosol per formare il complesso apoptosoma. Questo stimola la caspasi-9, che a sua volta attiva le caspasi effettrici. Il mitocondrio rilascia anche una proteina chiamata Smac/DIABLO nel citosol. Smac/DIABLO promuove indirettamente l’apoptosi bloccando gli effetti di un gruppo di proteine anti-apoptotiche chiamate proteine inibitrici dell’apoptosi (IAPs).
Le proteine anti-apoptotiche Bcl-2 e Bcl-XL inibiscono il rilascio del citocromo c, mentre Bax, Bak e Bid, tutte proteine pro-apoptotiche, promuovono il suo rilascio dai mitocondri. Il citocromo c e la deossiadenosina trifosfato (dATP) si legano all’APAF-1 per formare un complesso multimerico che recluta e attiva la pro-caspasi-9, una proteasi esecutrice mediatrice dell’apoptosi che a sua volta attiva la cascata delle caspasi, con conseguente apoptosi cellulare. Durante questo processo, le caspasi-2, caspasi-8, caspasi-9 e caspasi-10 sono coinvolte nell’inizio dell’apoptosi. Le caspasi-3, caspasi-6 e caspasi-7 sono coinvolte nell’apoptosi. La caspasi-3 e la caspasi-7 regolano l’inibizione della riparazione del DNA e iniziano la degradazione del DNA. Inoltre, la caspasi-6 regola la disintegrazione della lamina e del citoscheletro.
Percorso intrinseco di apoptosi in fisiopatologia
La maggior parte delle terapie chemioterapiche e mirate al cancro uccidono le cellule tumorali attraverso la generazione di segnalazione pro-morte che avvia il percorso intrinseco apoptotico di morte cellulare programmata. Il punto di non ritorno nella cascata apoptotica è la permeabilizzazione della membrana esterna mitocondriale (MOMP); una volta avvenuta, la permeabilizzazione mitocondriale porta alla formazione di un apoptosoma, che facilita l’attivazione delle caspasi e successivamente innesca le altre caratteristiche della morte cellulare apoptotica. La decisione cellulare di iniziare la MOMP è controllata da un delicato equilibrio tra le molecole pro- e anti-apoptotiche della famiglia BCL-2.
Una delle ragioni della resistenza alla chemioterapia è il fallimento delle cellule tumorali di andare in apoptosi a causa di difetti nella via apoptotica intrinseca (ad esempio, cambiamenti nella p53). Nonostante i significativi miglioramenti nel trattamento, i tassi di cura per molti tumori rimangono subottimali. L’aumento della chemioterapia citotossica ha portato a una terapia curativa per un sottogruppo di tumori, anche se la resistenza intrinseca al trattamento è difficile da prevedere per i singoli pazienti. L’ondata di terapie a bersaglio molecolare si è concentrata sulle mutazioni che attivano i farmaci ed è quindi limitata a specifici sottogruppi di pazienti. La via mitocondriale intrinseca dell’apoptosi rappresenta un bersaglio promettente per le nuove terapie, e colpire con successo questa via ha il potenziale di alterare il panorama terapeutico per una varietà di tumori.