Introducción
Las proteínas ß amiloide y tau fosforilada son sellos patológicos de la enfermedad de Alzheimer (EA) que se acumulan y se propagan de forma predecible a través de las redes neuronales distribuidas, causando anormalidades metabólicas progresivas, lesiones neuronales y muerte celular. Las neuroimágenes facilitan una evaluación detallada de estos cambios patológicos en los pacientes que se someten a un estudio de deterioro cognitivo. Anteriormente, el diagnóstico definitivo de la EA sólo era posible mediante la observación postmortem y la estadificación neuroanatómica de estos agregados proteicos. Sin embargo, los recientes avances en el campo de las imágenes moleculares permiten visualizar los depósitos de amiloide y tau en el cerebro humano vivo y nos han acercado a un diagnóstico definitivo in vivo de la EA.
Imagen estructural
Las directrices de la Academia Americana de Neurología (AAN) para la evaluación diagnóstica de las personas con problemas cognitivos1 recomiendan la realización de imágenes estructurales del cerebro con TC o RM sin contraste en cualquier persona con una historia clínica positiva y cambios cognitivos objetivos. En este contexto, el papel principal de las imágenes cerebrales es descartar lesiones estructurales no degenerativas, el 5% de las cuales pueden no ser evidentes en la historia clínica o la exploración física2 y son potencialmente tratables. La AAN también recomienda excluir clínicamente la demencia vascular (VaD), la demencia con cuerpos de Lewy (DLB) y la demencia frontotemporal (FTD). Las imágenes estructurales pueden mejorar la certeza diagnóstica y cambiar el diagnóstico clínico en el 19% al 28%, así como el manejo del 15% de los casos clínicos.2
Las modalidades de imagen de elección al evaluar la atrofia estructural son la TC y la RM. Los patrones de atrofia específicos de la enfermedad se han descrito y validado exhaustivamente mediante estas modalidades. Aunque la atrofia es observable en la TC, la resolución espacial intrínsecamente más baja y el contraste inferior entre la materia gris y la blanca hacen que se pierdan observaciones sutiles potencialmente útiles sobre los cambios neurodegenerativos. Por lo tanto, los médicos a menudo confían en la RM para evaluar el patrón y la gravedad de los cambios estructurales, para descartar las causas no neurodegenerativas del deterioro cognitivo y para evaluar la gravedad y el alcance de los cambios en la sustancia blanca, como se explica con más detalle a continuación.
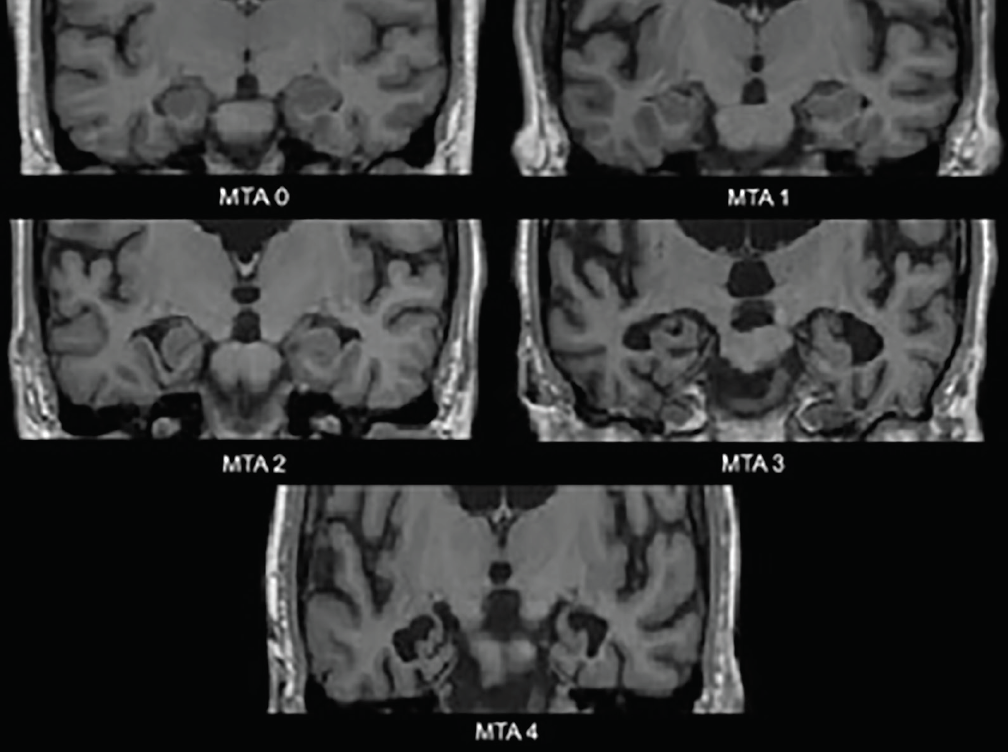
Los primeros cambios neurodegenerativos en la EA se producen en los lóbulos temporales mediales: el hipocampo, la corteza entorrinal y el giro parahipocampal. A medida que se produce la atrofia, la fisura coroidea y el cuerno temporal del ventrículo lateral se dilatan; estos cambios se evalúan mejor en el plano coronal. Una escala de evaluación visual comúnmente utilizada, basada en estas estructuras -la escala de atrofia temporal medial (MTA) (Figura 1)- ha sido validada clínica3 y neuropatológicamente4. Varios estudios que utilizan esta escala o las estructuras que evalúa han mostrado una capacidad significativa para discriminar a las personas con EA de los controles emparejados por edad4 o de aquellos con DCL4 o deterioro cognitivo vascular (DCV).5 La atrofia temporal medial también es predictiva de la conversión del deterioro cognitivo leve amnésico (DCL) a la demencia de Alzheimer.3
Haga clic para ver más grande
Figura 1. Calificaciones de la escala de atrofia temporal medial (MTA). MTA 0-ninguna o mínima separación de la fisura coroidea; MTA 1-ampliación sutil de la fisura coroidea; MTA 2-ampliación adicional de la fisura coroidea que se fusiona con el cuerno temporal del ventrículo lateral, disminución leve de la altura de la formación del hipocampo; MTA 3-disminución moderada de la altura de la formación del hipocampo, ampliación prominente del cuerno temporal del ventrículo lateral; MTA 4-disminución severa de la altura de la formación del hipocampo, ampliación prominente del cuerno temporal del ventrículo lateral.
Los síndromes de demencia suelen presentar patrones de atrofia canónicos6 que se corresponden con los síntomas cognitivos y conductuales; sin embargo, las estructuras implicadas pueden solaparse. Por ejemplo, tanto la EA como la DCL muestran una afectación del lóbulo temporal medial, pero varios estudios de imagen y neuropatológicos han demostrado que, en relación con la EA, la DCL tiene una predilección significativamente menor por las estructuras temporales mediales4,5 (Figura 2).
Haga clic para ver más grande
Figura 2. Patrones de atrofia estructural en la enfermedad de Alzheimer (EA) (A), la demencia con cuerpos de Lewy (DCL) (B), la demencia frontotemporal (DF) (C) y la demencia vascular (D). La resonancia magnética coronal ponderada en T1 demuestra una atrofia cortical generalizada pero diferentes grados de atrofia del hipocampo en la EA (A) y la DCL (B). La resonancia magnética axial ponderada en T1 demuestra una atrofia de la DFTB con una predilección focal por las cortezas prefrontales medial y lateral (C). La resonancia magnética axial ponderada en T2 demuestra hipointensidades de la materia blanca (WMHs) confluentes de la tapa periventricular y del halo que implican la corona radiata y se extienden a la neocorteza prefrontal lateral, WMHs leves dispersos y WMHs del revestimiento ventricular subependimario y del septum pellucidum en VaD (D).
Tanto la EA como la degeneración lobar frontotemporal (DLFT) tienen regiones de atrofia que se superponen, incluyendo áreas de la corteza prefrontal, orbitofrontal e insular anterior, así como los lóbulos temporales anterior y medial.6 Sin embargo, la EA muestra una atrofia significativamente mayor en las cortezas parietal y occipital laterales, mientras que la FTLD muestra más atrofia en los lóbulos frontales (Figura 2).6
Es importante evaluar sistemáticamente todas las áreas cerebrales, señalando las regiones de atrofia estructural y de preservación anatómica para hacer el mejor uso de las imágenes diagnósticas. Recientemente, un amplio estudio multicéntrico evaluó la capacidad de 6 escalas de valoración visual para clasificar correctamente 186 casos patológicamente confirmados de EA, DCL y DFT.6 Este estudio demostró que la evaluación simultánea de múltiples regiones específicas de la enfermedad proporcionaba una discriminación significativamente mejor que si se centraba en una sola área.4
El daño vascular es una consideración importante a la hora de evaluar a los individuos con deterioro cognitivo. El cambio isquémico es común en el envejecimiento normal y se acelera por comorbilidades comunes (por ejemplo, hipertensión, diabetes e hipercolesterolemia). Las contribuciones vasculares al deterioro cognitivo suelen inferirse cuando se detectan hiperintensidades significativas en la sustancia blanca (WMH), accidentes cerebrovasculares corticales o lagunas estratégicamente localizadas en secuencias ponderadas en T2 o de recuperación de inversión atenuada por fluidos (FLAIR). Los accidentes cerebrovasculares lacunares de los ganglios basales y la HSM en el centro semiovale y la corona radiata son indicadores de daño isquémico crónico de los vasos pequeños (Figura 2). Por el contrario, los tapones periventriculares suelen tener un origen no isquémico y reflejan una gliosis subependimaria.7 Existen varias escalas de gravedad de la HM (por ejemplo, la escala de Fazekas)7 que pueden utilizarse en el ámbito clínico. Todos estos algoritmos de puntuación coinciden en que la HM confluente generalizada y los infartos lacunares subcorticales son, al menos en parte, factores que contribuyen al deterioro cognitivo.7
La imagen con tensor de difusión (DTI) es una nueva secuencia de RM sensible a la difusión del agua a través del tejido que evalúa la integridad de la sustancia blanca porque la arquitectura de los axones limita el flujo de agua. Las medidas de difusividad media y anisotropía fraccional se utilizan a menudo para caracterizar los cambios neurodegenerativos en la DTI. La primera mide la magnitud de la difusión y la segunda mide si el agua fluye preferentemente en una sola dirección, como se espera en los axones sanos. La disminución de la difusividad media y la reducción de la anisotropía fraccional se han notificado sistemáticamente tanto en poblaciones con DCL como con demencia de EA.8 Varios estudios también han identificado cambios en la ITD en individuos asintomáticos con riesgo de desarrollar EA9 , lo que indica que estos cambios se producen en una fase temprana del curso de la enfermedad. Además de evaluar la integridad de la sustancia blanca, la DTI puede utilizarse para modelar la conectividad estructural bruta entre las regiones corticales utilizando algoritmos de tractografía. Esto ha ganado terreno en el campo de la EA, ya que las pruebas sugieren que la patología amiloide y tau puede propagarse a través de las conexiones cerebrales.10 Aunque la DTI muestra potencial como medida de la integridad de la sustancia blanca, su sensibilidad al movimiento y otras restricciones técnicas limitan, al menos por el momento, su aplicación clínica.
Imagen funcional
El hipometabolismo cerebral se observa fácilmente en los trastornos neurodegenerativos y puede ayudar al diagnóstico diferencial. La función de las neuronas depende del oxígeno y la glucosa de la sangre, cuyo suministro se ve facilitado por la vasodilatación regional. La tomografía por emisión de positrones con 18F-fluorodeoxiglucosa (FDG-PET) refleja indirectamente el grado de actividad cortical y puede utilizarse fácilmente para visualizar los cambios neurodegenerativos aprovechando esta dependencia metabólica de la glucosa.
La firma cortical hipometabólica característica de la EA consiste en cambios tempranos en el cíngulo posterior (PCC); el precuneus; las cortezas temporal, parietal y, en etapas posteriores, frontal. Este patrón se manifiesta tempranamente, incluso de forma presintomática,11 y es clínicamente útil para distinguir la EA de la FTD.12 A diferencia de la EA, la firma metabólica de la FTD revela un hipometabolismo frontal, temporal anterior, ganglionar basal y talámico, con una relativa preservación de las cortezas de asociación posteriores.13 En situaciones clínicas inciertas, los médicos pueden utilizar estas firmas metabólicas para desambiguar la EA de la FTD.12 Del mismo modo, aunque la firma metabólica neocortical de la DCL puede tener un solapamiento confuso con la de la EA, los casos de DCL también pueden mostrar hipometabolismo occipital.14
El Centro de Servicios de Medicare &Medicaid considera que la FDG-PET es «razonable y necesaria» sólo para aquellos que reúnen los criterios tanto de EA como de DCL, en los que el estudio diagnóstico completo según las directrices de la AAN1 no ha establecido definitivamente una etiología.15
La RMN funcional (fMRI) también puede medir la actividad cerebral mediante el uso de secuencias que son sensibles a los cambios temporales en la hemoglobina oxigenada/desoxigenada debido a la actividad cerebral. De este modo, la fMRI está relacionada con la FDG-PET porque los cambios en la hemoglobina son impulsados por el metabolismo de la glucosa necesario para la actividad cerebral. Las dependencias temporales entre regiones pueden utilizarse para inferir la conectividad funcional (es decir, qué regiones se comunican activamente). En comparación con la FDG-PET, la fMRI ofrece una mejor resolución espacial, lo que permite un mapeo más preciso de la conectividad. Otra ventaja de la RMf es que puede adquirirse durante la realización de una tarea o en reposo. La primera identifica patrones de conectividad impulsados por estados activos específicos, mientras que la segunda identifica patrones de conectividad presentes en reposo conocidos como la red de modo por defecto (DMN). La DMN está definida por núcleos funcionales, el PCC y el precuneus, que se conectan a un subsistema medial dorsal y a un subsistema temporal medial.16 Se cree que la deposición de amiloide comienza en los núcleos funcionales de la DMN. Se sabe que la DMN se ve afectada en la EA.16 Por el contrario, la DMN no se ve afectada en la FTD, mientras que sí lo están las redes de saliencia o de atención.17 En la actualidad, la tecnología de fMRI se limita a aplicaciones de investigación, pero se están explorando activamente vías para desarrollar algoritmos relevantes de aplicación clínica.
Imagen molecular
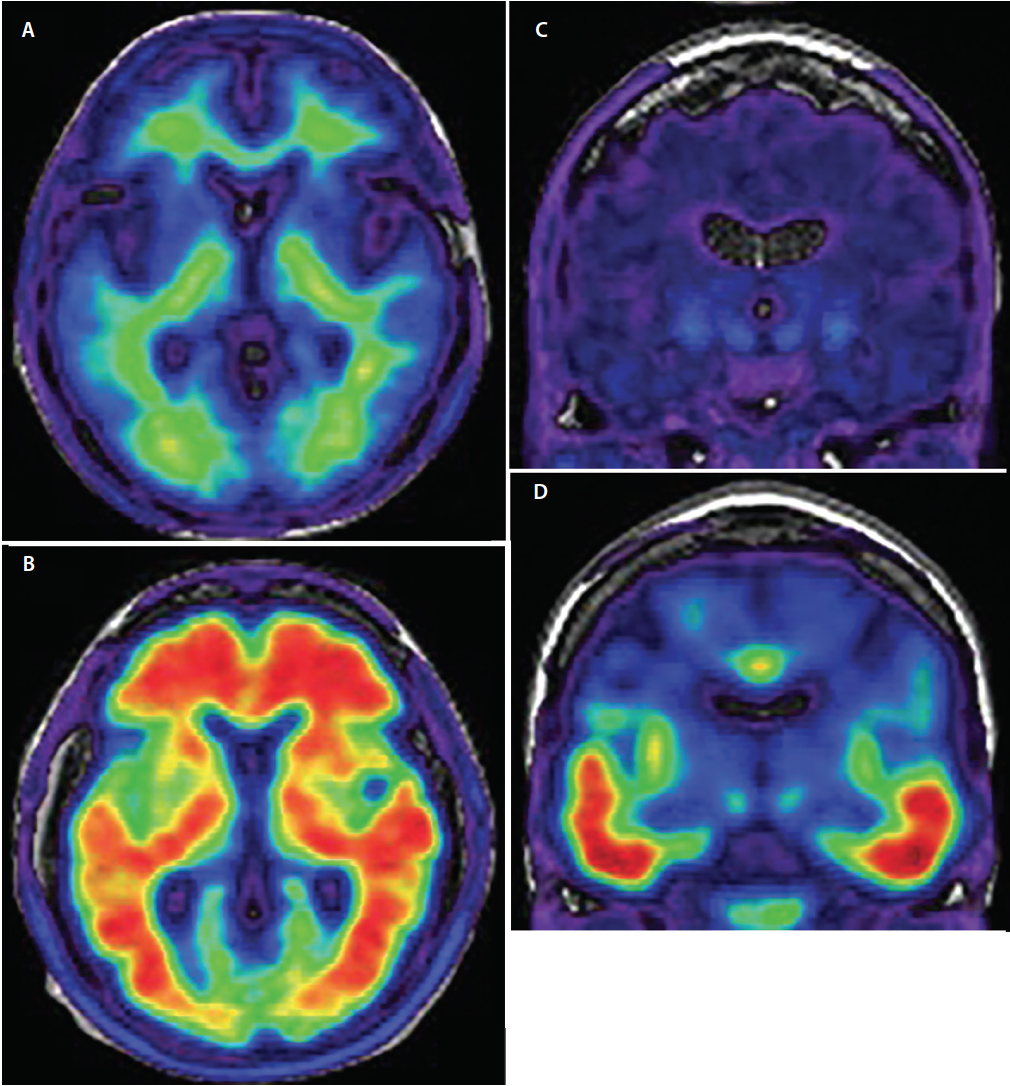
El criterio estándar para un diagnóstico definitivo de la EA es el examen neuropatológico del cerebro postmortem. Incluso en manos de expertos en demencia, el diagnóstico clínico de la EA sólo alcanza la inquietante sensibilidad del 70,9% al 87,3% y la especificidad del 44,3% al 70,8%.18 Lo más pertinente para mejorar la precisión del diagnóstico clínico de la EA ha sido el desarrollo de trazadores de imágenes PET radiomarcados con afinidad específica por los agregados de ß amiloide y tau fosforilados relacionados con la EA (Figura 3), que permiten la demostración in vivo de la neuropatología subyacente (Tabla) que antes sólo era posible postmortem.
Haga clic para ver más grande
Figura 3. TEP-amiloide axial demostrando exploraciones con amiloide negativo (A) y con amiloide positivo (B). En las imágenes negativas de amiloide (A) la señal del radiotrazador muestra una baja intensidad y se limita a la unión inespecífica de la materia blanca. La unión entre materia gris y blanca está preservada. En las imágenes positivas de amiloide (B) la señal del radiotrazador es de alta intensidad y se extiende difusamente en las regiones corticales de la materia gris oscureciendo la unión de la materia gris y blanca. Tau-PET coronal demostrando exploraciones tau negativas (C) y tau positivas (D). En las exploraciones negativas para tau hay una señal de radiotrazador mínima e inespecífica de baja intensidad en las regiones temporal media, cerebro anterior basal y ganglios basales. En las exploraciones positivas para tau (D) la señal del radiotrazador es de mayor intensidad y afecta a las cortezas temporales inferior y lateral siguiendo la trayectoria conocida de progresión de los ovillos neurofibrilares.
Tomografía por emisión de positrones de amiloide
El amiloide y, más recientemente, las imágenes de tau han revolucionado nuestra capacidad de visualizar la patología de la EA incluso en fases presintomáticas.19 Los trazadores de imágenes PET de amiloide son válidos y fiables para detectar la patología de la EA in vivo en varias etapas de la enfermedad. Estos trazadores de imágenes se unen a motivos conformacionales específicos de la proteína amiloide y dan lugar a una captación del trazador que está estrechamente correlacionada con la deposición de amiloide postmortem.20
En comparación con la tau, la patología amiloide comienza en un patrón regional de distribución más difusa que se extiende desde las porciones basales de los lóbulos frontal, temporal y occipital hasta las áreas de asociación neocortical dorsal, con una afectación sólo en la fase tardía de las cortezas sensoriales y motoras primarias.21 Las áreas más notables de captación del trazador son las corticales frontal, parietal y temporal lateral (Figura 3), con una captación de 1,5 a 2 veces mayor en las personas con EA frente a los grupos de control22 o aquellos con otras demencias.23
Las imágenes PET de amiloide se evalúan para la captación del trazador cortical en las áreas características de la distribución de amiloide de la EA. Las exploraciones positivas muestran la pérdida de la distinción entre materia gris y blanca a medida que la captación del trazador se extiende a la neocorteza. Las exploraciones negativas conservan la distinción entre materia gris y blanca, mostrando sólo la unión fuera del objetivo de la materia blanca.24 Los patrones de imágenes PET de amiloide pueden predecir de forma fiable el deterioro cognitivo tanto en personas sanas como en aquellas con DCL.20
El 18F-florbetapir, el 18F-florbetaben y el 18F-flutemetamol están aprobados por la Administración de Alimentos y Medicamentos (FDA) para su uso clínico.20 Sin embargo, las compañías de seguros no han adoptado la tecnología, debido al gasto y al riesgo de sobreutilización clínica en ausencia de terapias modificadoras de la enfermedad y de un coste-beneficio establecido. Un meta-análisis informó de una sensibilidad del 95% y una especificidad del 57% de una PET amiloide positiva para predecir la conversión de DCL a EA.25
La relación incierta entre el coste, el riesgo y el beneficio ha llevado al desarrollo de criterios de uso apropiado para las imágenes amiloides, atribuyendo la necesidad de la evaluación de PET amiloide a las personas con DCL inexplicable, presentaciones atípicas de EA y demencia de inicio temprano.26 Los expertos también han identificado indicaciones inapropiadas para la obtención de imágenes amiloides que incluyen la evaluación aislada de problemas cognitivos antes de un estudio clínico, cognitivo, de laboratorio y de neuroimagen estructural completo; en ausencia de un deterioro cognitivo objetivo; en caso de alta probabilidad de EA (es decir, en ausencia de un equilibrio clínico); y para la estadificación de la gravedad de la demencia.
El estudio de demencia por imágenes-evidencia para la exploración amiloide (IDEAS) -un estudio en curso del Centro de Servicios de Medicare y Medicaid- está validando actualmente los criterios de uso apropiado y evaluando el impacto del estado amiloide determinado por la PET en la gestión de la enfermedad y los resultados a largo plazo para los beneficiarios de Medicare con DCL o presentaciones atípicas. El análisis provisional reveló que la integración de la PET de amiloide en la evaluación clínica dio lugar a cambios en el tratamiento de la enfermedad en el 60,2% de las personas con DCL y en el 63,5% de las personas con demencia.27 Los resultados a largo plazo aún se están determinando.
Tomografía de emisión de positrones de Tau
Tau es la segunda proteína que se deposita en el cerebro de las personas con EA. Actualmente se están desarrollando trazadores de imágenes Tau PET que ya son prometedores. Al igual que la PET de amiloide, los trazadores de PET de tau se dirigen a motivos conformacionales concretos de la tau fosforilada. La especificidad de los trazadores de tau se ha validado postmortem,28 y la señal observada en la PET de tau coincide estrechamente con la distribución anatómica de los ovillos neurofibrilares que se utiliza actualmente para el diagnóstico neuropatológico de la EA.29 Las primeras etapas de la patología de tau que pueden visualizarse con la PET de tau son los depósitos de ovillos neurofibrilares en la corteza entorrinal y el hipocampo. A continuación, los depósitos de tau son detectables en la corteza temporal inferior y lateral (Figura 3), seguidos de la corteza parietal y occipital y, por último, de la corteza frontal, de acuerdo con la bien establecida estadificación patológica de Braak y Braak de la deposición de tau en el cerebro.21 Los trazadores de tau no están exentos de limitaciones diagnósticas y se están desarrollando para definir mejor su función clínica. Sin embargo, dado que la unión del trazador tau cambia dinámicamente a lo largo de todo el curso clínico de la EA, esta modalidad de imagen probablemente desempeñará un papel importante en la estadificación de la gravedad de la enfermedad in vivo.30
Direcciones futuras
Dado que la patología de la EA puede detectarse fácilmente hasta 20 años antes del diagnóstico de la demencia19 y sigue una distribución por etapas predecible, la investigación se centra ahora en la detección presintomática temprana y en la mejora de la precisión diagnóstica mediante el uso de biomarcadores (Tabla).31 Un marco de investigación propuesto de amiloide-tau-neurodegeneración (ATN) centrado en la detección de 3 cambios de biomarcadores en el cerebro pronostica la relevancia clínica de las imágenes multimodales.31 El sistema de clasificación ATN es agnóstico respecto a los síndromes clínicos y capta todo el espectro de enfermedades neurodegenerativas a través de una lente de biomarcadores de EA. Tras un mayor desarrollo y perfeccionamiento, se espera que la NTA, a través de la categorización objetiva de biomarcadores in vivo, mejore drásticamente la precisión del diagnóstico clínico y mejore la calidad de la atención.
Conclusión
La neuroimagen ofrece información única sobre la etiología subyacente del deterioro cognitivo y facilita la orientación de los pacientes y las familias a través de una experiencia temerosa e incierta. Todas las enfermedades neurodegenerativas muestran una importante heterogeneidad clínica y, antes de los últimos avances en imagen molecular, ninguna podía diagnosticarse definitivamente antes de la muerte. La moderna tecnología de imagen cerebral es capaz de detectar multitud de epifenómenos que reflejan la neuropatología subyacente, algunos de los cuales son bastante próximos al origen de la enfermedad. En la actualidad, el diagnóstico clínico más fiable se basa en la integración de la historia clínica, las observaciones del médico, la exploración física, el estudio médico, las pruebas neuropsicológicas y la interpretación informada del diagnóstico por neuroimagen. A medida que se perfeccionen los biomarcadores de la NTA, los diagnósticos clínicos se realizarán antes y de forma más definitiva en los pacientes vivos, dependerán menos de la clasificación de los síntomas y proporcionarán oportunidades para una intervención terapéutica más temprana que pueda alterar la trayectoria de la enfermedad neurodegenerativa.
1. Knopman DS, DeKosky ST, Cummings JL, et al. Parámetro de práctica: diagnóstico de la demencia (una revisión basada en la evidencia). Informe del Subcomité de Estándares de Calidad de la Academia Americana de Neurología. Neurología. 2001;56(9):1143-1153.
2. Chui H, Zhang Q. Evaluation of dementia: a systematic study of the usefulness of the American Academy of Neurology’s practice parameters. Neurology. 1997;49(4):925-935.
3. Apostolova LG, Thompson PM, Green AE, et al. 3D comparison of low, intermediate, and advanced hippocampal atrophy in MCI. Hum Brain Mapp. 2010;31(5):786-797.
4. Harper L, Fumagalli GG, Barkhof F, et al. MRI visual rating scales in the diagnosis of dementia: evaluation in 184 post-mortem confirmed cases. Brain. 2016;139(Pt 4):1211-1225.
5. Burton EJ, Barber R, Mukaetova-Ladinska EB, et al. La atrofia del lóbulo temporal medial en la RMN diferencia la enfermedad de Alzheimer de la demencia con cuerpos de Lewy y el deterioro cognitivo vascular: un estudio prospectivo con verificación patológica del diagnóstico. Brain. 2009;132(Pt 1):195-203.
6. Harper L, Bouwman F, Burton EJ, et al. Patterns of atrophy in pathologically confirmed dementias: a voxelwise analysis. J Neurol Neurosurg Psychiatry. 2017;88(11):908-916.
7. Fazekas F, Kleinert R, Offenbacher H, et al. Correlatos patológicos de las hiperintensidades de señal de la materia blanca de la RMN incidental. Neurology. 1993;43(9):1683-1689.
8. Huang J, Friedland RP, Auchus AP. Diffusion tensor imaging of normal-appearing white matter in mild cognitive impairment and early Alzheimer disease: preliminary evidence of axonal degeneration in the temporal lobe. AJNR Am J Neuroradiol. 2007;28(10):1943-1948.
9. Selnes P, Fjell AM, Gjerstad L, et al. White matter imaging changes in subjective and mild cognitive impairment. Alzheimers Dement. 2012;8(5 Suppl):S112-S121.
10. Jacobs HIL, Hedden T, Schultz AP, et al. Structural tract alterations predict downstream tau accumulation in amyloid-positive individuals. Nat Neurosci. 2018;21(3):424-431.
11. Apostolova LG, Thompson PM, Rogers SA, et al. Mapeo guiado por características superficiales de los cambios metabólicos cerebrales en ancianos cognitivamente normales y con deterioro leve. Mol Imaging Biol. 2010;12(2):218-224.
12. Foster NL, Heidebrink JL, Clark CM, et al. FDG-PET mejora la precisión en la distinción de la demencia frontotemporal y la enfermedad de Alzheimer. Brain. 2007;130(Pt 10):2616-2635.
13. Ishii K, Sakamoto S, Sasaki M, et al. Metabolismo cerebral de la glucosa en pacientes con demencia frontotemporal. J Nucl Med. 1998;39(11):1875-1878.
14. Mosconi L, Tsui WH, Herholz K, et al. Multicenter standardized 18F-FDG PET diagnosis of mild cognitive impairment, Alzheimer’s disease, and other dementias. J Nucl Med. 2008;49(3):390-398.
15 . Centros para Medicare & Medicaid Serivces. Memo de decisión para la tomografía por emisión de positrones (FDG) y otros dispositivos de neuroimagen para la sospecha de demencia (CAG-00088R)https://www.cms.gov/medicare-coverage-database/details/nca-decision-memo.aspx?NCAId=104. Consultado el 4 de mayo de 2019.
16. Buckner RL, Sepulcre J, Talukdar T, et al. Hubs corticales revelados por la conectividad funcional intrínseca: mapeo, evaluación de la estabilidad y relación con la enfermedad de Alzheimer. J Neurosci. 2009;29(6):1860-1873.
17. Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Las enfermedades neurodegenerativas se dirigen a las redes cerebrales humanas a gran escala. Neuron. 2009;62(1):42-52.
18. Beach TG, Monsell SE, Phillips LE, Kukull W. Precisión del diagnóstico clínico de la enfermedad de Alzheimer en los Centros de Enfermedades de Alzheimer del Instituto Nacional sobre el Envejecimiento, 2005-2010. J Neuropathol Exp Neurol. 2012;71(4):266-273.
19. Jagust W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat Rev Neurosci. 2018;19(11):687-700.
20. Rice L, Bisdas S. El valor diagnóstico de la FDG y la PET amiloide en la enfermedad de Alzheimer: una revisión sistemática. Eur J Radiol. 2017;94:16-24.
21. Braak H, Braak E. Estadificación neuropatológica de los cambios relacionados con el Alzheimer. Acta Neuropathol. 1991;82(4):239-259.
22. Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55(3):306-319.
23. Ossenkoppele R, Jansen WJ, Rabinovici GD, et al. Prevalencia de la positividad de la PET amiloide en los síndromes de demencia: un meta-análisis. JAMA. 2015;313(19):1939-1949.
24. Minoshima S, Drzezga AE, Barthel H, et al. SNMMI procedure standard/EANM Practice Guideline for Amyloid PET Imaging of the Brain 1.0. J Nucl Med. 2016;57(8):1316-1322.
25. Ma Y, Zhang S, Li J, et al. Precisión predictiva de las imágenes amiloides para la progresión del deterioro cognitivo leve a la enfermedad de Alzheimer con diferentes longitudes de seguimiento: un meta-análisis. . Medicine (Baltimore). 2014;93(27):e150.
26. Johnson KA, Minoshima S, Bohnen NI, et al. Criterios de uso apropiado para la PET amiloide: un informe del Grupo de Trabajo de Imagen Amiloide, la Sociedad de Medicina Nuclear e Imagen Molecular, y la Asociación de Alzheimer. Alzheimers Dement. 2013;9(1):e1-e16.
27. Rabinovici GD, Gatsonis C, Apgar C, et al. Asociación de la tomografía de emisión de positrones amiloide con el cambio posterior en la gestión clínica entre los beneficiarios de medicare con deterioro cognitivo leve o demencia. JAMA. 2019;321(13):1286-1294.
28. Aguero C, Dhaynaut M, Normandin MD, et al. Validación autorradiográfica del nuevo trazador tau PET -MK-6240 en tejido cerebral humano postmortem. Acta Neuropathol Commun. 2019;7(1):37.
29. Chien DT, Bahri S, Szardenings AK, et al. Primeros resultados clínicos de imágenes PET con el nuevo radioligando PHF-tau -T807. J Alzheimers Dis. 2013;34(2):457-468.
30. Wang L, Benzinger TL, Su Y, et al. Evaluation of tau imaging in staging Alzheimer disease and revealing interactions between beta-amyloid and tauopathy. JAMA Neurol. 2016;73(9):1070-1077.
31. Jack CR, Jr, Bennett DA, Blennow K, et al. Marco de investigación NIA-AA: Hacia una definición biológica de la enfermedad de Alzheimer. Alzheimers Dement. 2018;14(4):535-562.
MRA y DS informan que no han revelado nada.
LGA ha sido miembro del consejo asesor de Eli Lilly.